ANOVA
Last updated on 2026-06-15 | Edit this page
Overview
Questions
- How do you perform an ANOVA?
- What even is ANOVA?
Objectives
- Explain how to run an analysis of variance on models
- Explain the requisites for runnin an ANOVA
- Explain what an ANOVA is
What is ANOVA
ANOVA (ANalysis Of VAriance) is a statistical method used to compare the means of three or more groups in order to determine if there is a significant difference between them.
Or, more simply: Are the means of all the groups equal?
An example
Studying the length of penguin flippers, we notice that there is a difference between the average length between three different species of penguins:
R
library(tidyverse)
library(palmerpenguins)
penguins |>
group_by(species) |>
summarise(mean_flipper_length = mean(flipper_length_mm, na.rm = TRUE))
OUTPUT
# A tibble: 3 × 2
species mean_flipper_length
<fct> <dbl>
1 Adelie 190.
2 Chinstrap 196.
3 Gentoo 217.If we only had two groups, we would use a t-test to determine if
there is a significant difference between the two groups. But here we
have three. And when we have three, or more, we use the ANOVA-method, or
rather the aov() function:
R
aov(flipper_length_mm ~ species, data = penguins) |>
summary()
OUTPUT
Df Sum Sq Mean Sq F value Pr(>F)
species 2 52473 26237 594.8 <2e-16 ***
Residuals 339 14953 44
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
2 observations deleted due to missingnessWe are testing if there is a difference in flipper_length_mm when we explain it as a function of species. Or, in other words, we analyse how much of the variation in flipper length is caused by variation between the groups, and how much is caused by variation within the groups. If the difference between those two parts of the variation is large enough, we conclude that there is a significant difference between the groups.
In this case, the p-value is very small, and we reject the NULL-hypothesis that there is no difference in the variance between the groups, and conversely that we accept the alternative hypothesis that there is a difference.
Are we allowed to run an ANOVA?
There are some conditions that needs to be fullfilled.
- The observations must be independent.
In this example that we can safely assume that the length of the flipper of a penguin is not influenced by the length of another penguin.
- The residuals have to be normally distributed
Typically we also test if the data is normally distributed. Let us look at both:
Is the data normally distributed?
R
penguins |>
ggplot(aes(x=flipper_length_mm)) +
geom_histogram() +
facet_wrap(~species)
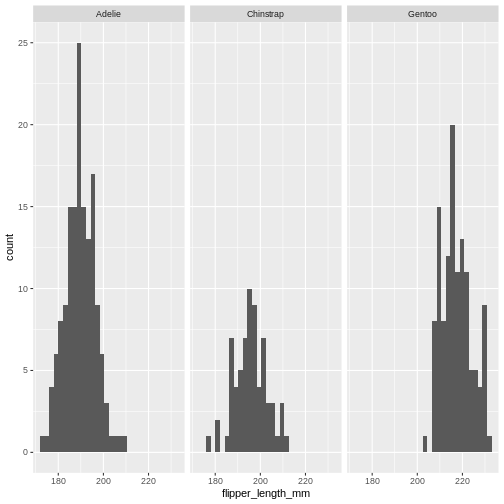 That looks reasonable.
That looks reasonable.
And the residuals?
R
aov(flipper_length_mm ~ species, data = penguins)$residuals |>
hist(.)
ERROR
Error:
! object '.' not foundThat looks fine - if we want a more specific test, those exists, but will not be covered here.
- Homoskedacity
A weird name, it simply means that the variance in the different groups are more or less the same. We can calculate the variance and compare:
R
penguins |>
group_by(species) |>
summarise(variance = var(flipper_length_mm, na.rm = TRUE))
OUTPUT
# A tibble: 3 × 2
species variance
<fct> <dbl>
1 Adelie 42.8
2 Chinstrap 50.9
3 Gentoo 42.1Are the variances too different? As a rule of thumb, we have a problem if the largest variance is more than 4-5 times larger than the smallest variance. This is OK for this example.
If there is too large difference in the size of the three groups, smaller differences in variance can be problematic.
There are probably more than than three tests for homoskedacity, here are three:
Fligner-Killeen test:
R
fligner.test(flipper_length_mm ~ species, data = penguins)
OUTPUT
Fligner-Killeen test of homogeneity of variances
data: flipper_length_mm by species
Fligner-Killeen:med chi-squared = 0.69172, df = 2, p-value = 0.7076Bartlett’s test:
R
bartlett.test(flipper_length_mm ~ species, data = penguins)
OUTPUT
Bartlett test of homogeneity of variances
data: flipper_length_mm by species
Bartlett's K-squared = 0.91722, df = 2, p-value = 0.6322Levene test:
R
library(car)
OUTPUT
Loading required package: carDataOUTPUT
Attaching package: 'car'OUTPUT
The following object is masked from 'package:dplyr':
recodeOUTPUT
The following object is masked from 'package:purrr':
someR
leveneTest(flipper_length_mm ~ species, data = penguins)
OUTPUT
Levene's Test for Homogeneity of Variance (center = median)
Df F value Pr(>F)
group 2 0.3306 0.7188
339 For all three tests - if the p-value is >0.05, there is a significant difference in the variance - and we are not allowed to use the ANOVA-method. In this case we are on the safe side.
But where is the difference?
Yes, there is a difference between the average flipper length of the three species. But that might arise from one of the species having extremely long flippers, and there not being much difference between the two other species.
So we do a posthoc analysis to confirm where the differences are.
The most common is the tukeyHSD test, HSD standing for “Honest
Significant Differences”. We do that by saving the result of our
ANOVa-analysis and run the TukeyHSD() function on the
result:
R
aov_model <- aov(flipper_length_mm ~ species, data = penguins)
TukeyHSD(aov_model)
OUTPUT
Tukey multiple comparisons of means
95% family-wise confidence level
Fit: aov(formula = flipper_length_mm ~ species, data = penguins)
$species
diff lwr upr p adj
Chinstrap-Adelie 5.869887 3.586583 8.153191 0
Gentoo-Adelie 27.233349 25.334376 29.132323 0
Gentoo-Chinstrap 21.363462 19.000841 23.726084 0We get the estimate of the pair-wise differences and lower and upper 95% confidence intervals for those differences.
Try it on your own
The iris dataset, build into R, contains measurements of
three different species of the iris-family of flowers.
Is there a significant difference between the
Sepal.Width of the three species?
R
aov(Sepal.Width ~Species, data = iris) |>
summary()
OUTPUT
Df Sum Sq Mean Sq F value Pr(>F)
Species 2 11.35 5.672 49.16 <2e-16 ***
Residuals 147 16.96 0.115
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Try it on your own (continued)
The result only reveal that there is a difference. Or rather that we can reject the hypothesis that there is no difference.
Which species differ?
R
aov(Sepal.Width ~Species, data = iris) |>
TukeyHSD()
OUTPUT
Tukey multiple comparisons of means
95% family-wise confidence level
Fit: aov(formula = Sepal.Width ~ Species, data = iris)
$Species
diff lwr upr p adj
versicolor-setosa -0.658 -0.81885528 -0.4971447 0.0000000
virginica-setosa -0.454 -0.61485528 -0.2931447 0.0000000
virginica-versicolor 0.204 0.04314472 0.3648553 0.0087802- Use
.mdfiles for episodes when you want static content